

The US Food and Drug Administration is tasked with protecting consumers from unsafe food and medical products. In new research, Parker Rogers finds that FDA regulations can not only hinder innovation and competition but often the improvements to product safety they are supposed to encourage.
Effective regulation of new technologies can safeguard consumers from harm, but excessive regulation can stifle the development of life-saving technologies. The Covid-19 pandemic highlighted this delicate balance, with FDA regulations delaying the rapid commercialization of newly developed vaccines, diagnostic tests, and ventilators. However, this balancing act is not unique to times of crisis but a constant challenge in the regulation of the $2.8 trillion worth of goods overseen each year by the US Food and Drug Administration (FDA). Finding better ways to strike this balance is essential for ensuring consumer safety and supporting innovation in health care markets.
Regulation can take many forms. The FDA currently uses three regulatory classes to oversee approximately 5,500 medical device types, like mammography machines or spinal implants. Class III is the most stringent class and requires expensive clinical trials demonstrating the safety and effectiveness of devices. Brand-name drugs face similar regulatory requirements. Class II requires device manufacturers to prove that their product is similar to an existing technology. Genetically modified crops and foods, generic drugs, and tobacco products are regulated similarly. Lastly, Class I requires no testing, but devices in this class are not as protected from legal liability risk related to a product’s design. This lack of legal protection may encourage inventors to create safer products.
The FDA sometimes shifts medical device types to a lower class—a process called down-classification—based on the type’s safety profile. The FDA determines a medical device type’s safety profile based on adverse event reports related to its use, clinical trial data from regulatory submissions, and other scientific sources of safety information. These episodes of deregulation, which can reduce the upfront business costs of approving a new device by as much as $50 million, provide an opportunity to study the relative costs and benefits of these three regulatory approaches.
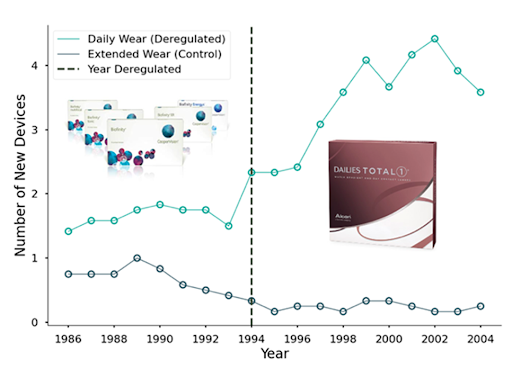
One example of down-classification occurred in 1994 when the FDA down-classified daily-wear soft contact lenses from Class III to Class II. The FDA revealed in the 1993 regulatory panel meeting announcing the down-classification that it had determined through an analysis of safety information in regulatory submissions that these products were safe enough for down-classification almost a decade earlier, but bureaucratic hurdles had delayed the process. After down-classification, the number of new daily-wear soft contact lenses increased sharply, while the number of new Class III extended-wear soft contact lenses remained steady.
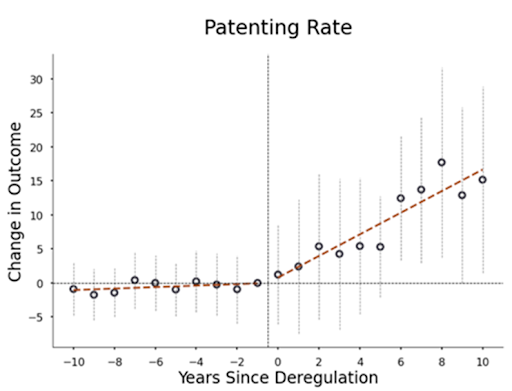
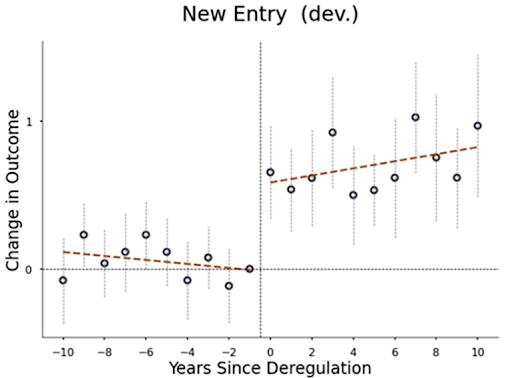
In my recent paper, I systematically analyze all such down-classifications from 1980-2015. When device types were down-classified from Class III to II, the number of patents filed and FDA device submissions for that type increased by up to 400%. This increase suggests that approval costs play an important role in determining which research and development (R&D) projects companies pursue. Small companies and those with limited regulatory experience saw the largest increases in innovation, perhaps because they are more likely to have difficulty securing financing and navigating the complex approval process. Additionally, when device types were down-classified from Class III to II, they attracted 1,000% more new manufacturers, leading to increased competition and a roughly 35% decrease in the prices of medical procedures using these deregulated devices (e.g., spinal fusion procedures using spinal implants).
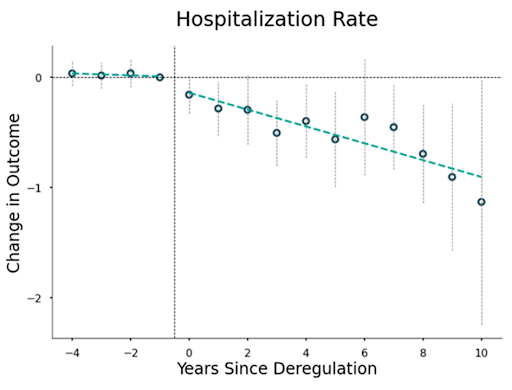
The benefits of down-classification generally outweigh the potential costs to safety. When device types were down-classified from Class III to II, the number of hospitalizations related to the use of these devices increased, but measures of mortality and life-threatening events did not change. On the other hand, when device types were down-classified from Class II to I, inventors mentioned product safety improvements more frequently in their patents, and the number of serious adverse events decreased.
This safety improvement may occur because deregulation increases the incentives for innovators to improve product safety. In the US, Class III devices, and to a lesser extent Class II devices (with special controls), are protected from product design lawsuits. After down-classification, innovators face greater legal liability for unsafe products.
Indeed, large companies, which cannot use bankruptcy to avoid paying significant damages in the event of litigation, tend to place the greatest emphasis on product safety after down-classification and experience the greatest reductions in adverse events. On the other hand, small companies, which can avoid worst-case damages through bankruptcy, do not increase their focus on product safety as much, resulting in smaller reductions in adverse events. These findings support the idea that legal liability is a key driver of product safety improvements after down-classification, although other factors may also be at play.
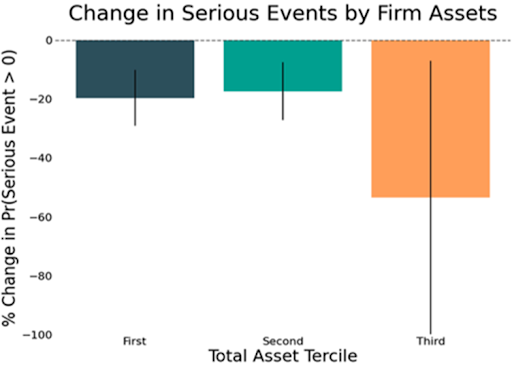
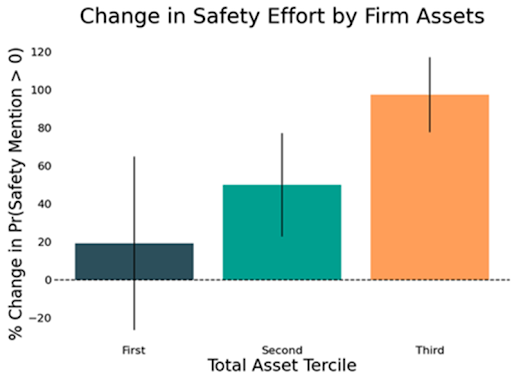
The way in which the FDA has determined Class II to I down-classifications suggests that the FDA could be managing the tradeoffs of deregulation more effectively. To make these decisions, in 1990, the FDA developed a crude measure of safety based on annual counts of deaths, hospitalizations, and other serious health events. Device types that fell below a threshold of adverse events were down-classified. This regulatory approach highlights two key issues. First, it ignores the potential impact of down-classification on innovation, market structure, and prices. Second, it turns out that the safety benefits of deregulation were greatest for Class II devices that the crude measure considered more dangerous (i.e., marginal) but were still down-classified. This finding suggests that overall product safety could have improved if unaffected Class II devices that were not deregulated had been down-classified and that the FDA is too cautious in its approach.
It is worth noting that it is possible that, on average, the benefits of deregulation outweigh the costs, but that for more dangerous (marginal) devices, the FDA’s regulatory framework may be efficient. However, the fact that more dangerous devices become even safer relative to less dangerous ones after down-classification challenges the FDA’s approach to Class II devices.
Overall, these findings suggest that the benefits of deregulating more Class II devices could outweigh the costs. The National Academy of Medicine has long criticized Class II regulations as inadequate—my results suggest Class I regulations are a compelling alternative. Additionally, it is possible that some current Class III devices may be worth down-classifying based on the benefits from past Class III to II down-classifications. More generally, the pattern observed in the FDA’s regulation of medical devices suggests that there may be more effective ways of determining regulatory standards for medical technologies.
The FDA could improve its regulatory processes by collaborating with external researchers and using experimentation as a tool. For example, the FDA could more readily allow external researchers to access internal data to examine its processes instead of requiring researchers to request data through the years-long FOIA process. Additionally, the FDA could follow the lead of institutions like the US Patent and Trademark Office and pursue a more hands-on approach that incorporates the use of randomized assignment of review staff to, for example, test for biases and inefficiencies in the approval process.
The FDA could go further by partnering with researchers to pilot new regulatory approaches and compare them to the current system using randomized evaluation, like how medical technology companies use experiments to demonstrate the safety and effectiveness of their products. For example, the FDA may be interested in studying the effects of streamlining the approval process on product safety and innovation, as it did through the FDA’s Breakthrough Therapy Designation. These partnerships would be mutually beneficial, as researchers could gain a deeper understanding of FDA processes, identify important research questions, and access internal data to improve how they measure outcomes. Meanwhile, the FDA would benefit from external help addressing key issues through rigorous randomized evaluation.
By incorporating experimentation and partnerships into its processes, the FDA can build a stronger evidence base for its decision-making and improve the effectiveness of its regulatory activities, ultimately ensuring that the FDA is held to the same high standards to which it holds innovators.